ClinicalTrials.gov FAQs
Last Updated: August 26, 2025 2:52:46 PM PDT
Give feedback
Expand section GENERAL QUESTIONS
Are there two ClinicalTrials.gov websites?
ClinicalTrials.gov has both a public site, for the public to view clinical trials, and a private user platform, “Protocol Registration and Results System (PRS), that is used by study team members to upload and maintain their clinical trial study record(s).
Public Site: https://clinicaltrials.gov/
PRS Site: https://register.clinicaltrials.gov
What is PROCoM?
PROCoM is a system, built by UCLA, to help the institutional Protocol Registration and Results System (PRS) Administrator manage UCSD’s PRS “problem” records. This system was used by UCSD to deliver messages regarding clinicaltrials.gov problem records to Responsible Parties and Record Owners, and emails generated from it were from RCIClinicalTrialsGov@PRSTracker.net. The Research Compliance and Integrity Office has since retired the use of this system as of May 2025, and now sends updates regarding problem records via the ctgov@ucsd.edu email address.
Can I add people to make changes to the record who are not the Responsible Party or the Record Owner?
The Record Owner and the Responsible Party, by default have access to the record and can add information or modify information in that record. The Record Owner or Responsible Party can grant access to additional users by using the record's “Access List.” Users on a record's “Access List” can update and edit a record.
To update the Access List, use the Edit Access List link on the Record Summary page and select the usernames of people who should have access to the record.
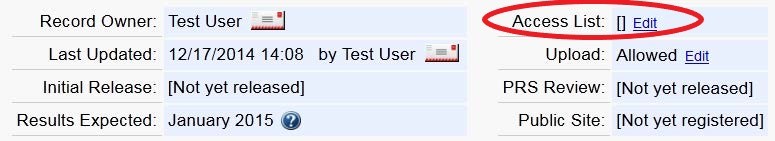
When users are added to an Access List, the record will be available through their Record List when they log into ClinicalTrials.gov. Users being added to the Access List must have a PRS account prior to being successfully added.
What does the Record Status mean?

Does ClinicalTrials.gov provide guidance to questions related to COVID-19?
Yes. Please see Responses to Top Questions from Responsible Parties Related to Coronavirus (COVID-19) (clinicaltrials.gov) (PDF)
What is the Primary Completion Date (PCD)?
The Primary Completion Date (PCD) is the date the final participant was examined or received an intervention for the purposes of final collection of data for the primary outcome, whether the study concluded according to the pre-specified protocol or was terminated. In the case of clinical studies with more than one primary outcome measure where data is collected over different Time Frames, this term refers to the date upon which data collection is complete from all enrolled participants for all of the primary outcomes.
Once the study has reached the PCD, the Responsible Party must update it from Anticipated to Actual in the ClinicalTrials.gov record within 30 days.
For Applicable Clinical Trials (ACT) per FDAAA Law and NIH-funded clinical trials, results will be due one year from the PCD. Because the federal due date for results hinges on the accurate and timely entry of the PCD, it is imperative that study teams understand the PCD definition and the requirements surrounding its update.
- What are some other dates that study teams might mistakenly enter as PCD?
- Study teams may mistakenly enter in the following incorrect dates as PCD:
- Date that enrollment closed
- Date the data was analyzed
- Date the protocol was closed with IRB
- Date of publication
- Date the protocol was terminated
- Study teams may mistakenly enter in the following incorrect dates as PCD:
Common Primary Completion Date (PCD) Questions and Answers
- If tissue samples (blood, biopsy, etc.) are collected from a participant per protocol to answer a Primary Outcome, is the PCD the date that the last sample was collected or the date that the last sample was analyzed?
-
- Per the PCD definition and the ClinicalTrials.gov FAQs, the PCD is the date that the last sample was collected from the last participant and NOT the date that it was assessed, analyzed, or interpreted.
- If participant survey responses are collected or telephone interviews are conducted with a participant per protocol to answer a Primary Outcome, how does this inform the PCD?
-
- Per the ClinicalTrials.gov FAQs, there are a broad range of data collection methods inferred in FDAAA Law, including examination via phone or electronic means. If the study employees survey responses or telephone interviews to answer a Primary Outcome, it would be legally permissible to use the date of the last participant’s survey conducted / received or the date of the last participant’s telephone interview, as applicable, as the PCD. The same would be true if the examination was via telephone or other electronic means. If the survey is via paper sent through the mail, it is permissible to use date of receipt in the mail as the date of data collection.
- What is the PCD for Terminated Trials?
-
- The PCD for Terminated Trials remains the date the final participant was examined or received an intervention for the purposes of final collection of data for the primary outcome. It is NOT the date upon which the trial was terminated. Bear in mind that if an ACT (or NIH-funded clinical trial) is terminated and the PCD occurred over a year ago, this means results are now late and an update of the PCD in the public record would indicate as such.
Expand section REGISTRATION QUESTIONS
How do I get a user account for the UCSD Protocol Registration and Results System (PRS) account to register my clinical trial and to post results?
Email ctgov@ucsd.edu and request a new user account. A new account will be set up for you and ClinicalTrials.gov will send you an email with your login username and temporary password. After you log in for the first time, make sure to change your password. To change your password go to Accounts> Change Password.
Who is the Responsible Party? What is the Responsible Party responsible for?
Each clinical trial registered on ClinicalTrials.gov must have a designated Responsible Party (RP). The RP is legally responsible for registering the clinical trial on ClinicalTrials.gov, ensuring the record is accurate and that the content is up-to-date. There can be only one RP for each clinical trial registered. The person identified as the Responsible Party for the Sponsor Organization account acts as the official contact person for that account and full contact information for that person must be listed in the Protocol Registration and Results System (PRS) account.
If a clinical trial is Industry Initiated/Sponsored, the Industry Sponsor must serve as the RP. Some institutions allow the Industry Sponsor to transfer the RP duties to the Principal Investigator; however, this is not permitted at UCSD. UCSD, as an entity, cannot be the RP. Clinical trials must list the Principal investigator or the Industry Sponsor as the RP. Use the Factsheet below to help determine the RP.
Factsheet: Determining the Responsible Party decision tree
NOTE: This differs from the IRB application which does list UCSD as the sponsor of Investigator Initiated Trials. But for purposes of ClinicalTrials.gov only, the Prinicpal Investigator is the Sponsor.
If our study is a multi-site study, do all the sites have to register on ClinicalTrials.gov?
Multi-site studies are NOT registered by individual sites. If this is a multi-site study, it must be registered only once, by the Responsible Party (IND/IDE holder or the person or organization who initiates the study and who has authority and control over the study) or its designated Principal Investigator (PI).
What is the organizations unique “protocol ID”?
The Unique Protocol ID is the number/name that the record will be titled until an NCT number is assigned. It is suggested you use either the UCSD IRB number (if already submitted to the IRB) or the UCSD Proposal Number.
What is a Secondary Identification?
If the clinical trial is funded in whole or in part by a U.S. Federal Government agency, the complete grant or contract number must be submitted as a Secondary Identification (ID). When entering a Secondary ID, select one of the following types:
- U.S. National Institutes of Health (NIH) Grant/Contract Award Number: In the Secondary ID field, include activity code, institute code, and 6-digit serial number. For example 026776.
- Other Grant/Funding Number: Identifier assigned by a funding organization other than the NIH (note, the name of the funding organization is also required).
- Registry Identifier: Number assigned by a clinical trial registry. For example, a registry that is part of the World Health Organization [WHO] Registry Network (note the name of the clinical trial registry is also required).
- EudraCT Number: Identifier assigned by the European Medicines Agency Clinical Trials Database (EudraCT).
- Other Identifier: Enter a brief description of the Identifier. For example, the name of organization that issued the Identifier.
UC San Diego encourages all Responsible Parties to enter the name of the funding organization and funding/grant number in the Secondary ID field regardless of the funding source.
What is a Collaborator?
A collaborator is any other organizations (as applicable) that are providing support. Support may include funding, design, implementation, data analysis or reporting. The Responsible Party is responsible for confirming all collaborators before including them on the ClinicalTrials.gov record.
UC San Diego encourages the Responsible Party to include all funding sources as a collaborator.
What is Individual Participant Data (IPD)?
This question asks whether there is a plan to make individual participant data (IPD) collected in this study, including data dictionaries, available to other researchers (typically after the end of the study).
For interventional studies, when "Undecided" is selected for the "Plan to Share IPD" data element, a NOTE now appears indicating that the International Committee of Medical Journal Editors (ICMJE) data sharing policy requires a "Yes" or "No" answer to this question. The "Plan to Share IPD" data element is optional in the PRS, but is required by the ICMJE as part of registration information for interventional studies. For more information, see the ICMJE data sharing recommendations.
What is the "annual due date?"
Each study record registered on ClinicalTrials.gov must be updated at least every twelve (12) months. This means the record is reveiwed and the verification date under "Study Status" is updated.
Are Expanded Access studies required to be registered on ClinicalTrials.gov?
The final rule clarifies that expanded access (EA) use of a drug, biological, or device product is not considered an "applicable clinical trial" (ACT) under the definition in 42 CFR 11.10 (81 FR 65009-10). Therefore, the submission of clinical trial registration and results information for EA use would not be required by 42 CFR 11.22 and 42 CFR 11.42.
However, if the responsible party for an ACT of an unapproved drug or an unlicensed biological product is both the sponsor of the ACT being registered and the manufacturer of the unapproved product, there is a requirement for a submission of a separate expanded access record containing details about how to obtain access to the investigational product.
Once an expanded access record has been created for a particular investigational product and a National Clinical Trial (NCT) number has been assigned, the responsible party must update the ACT(s) with that NCT number and provide that NCT number when submitting clinical trial registration information for any future ACT(s) studying the same investigational product. The NCT number for the expanded access record allows ClinicalTrials.gov to link the existing expanded access record to the study record for the clinical trial.
Are FDA Phase 1 studies required to be registered?
A phase 1 (e.g. includes the initial introduction of an investigational new drug into humans to determine the metabolism and pharmacologic actions of the drug in humans and the side effects associated with increasing doses) clinical trial does not meet the FDA definition of an Applicable Clinical Trial and therefore, generally, does not have to be registered.
Some clinical trials are classified as a ‘‘phase 0’’ (e.g., to refer to clinical trials that areexploratory in nature and are not designed to evaluate therapeutic or diagnostic intent). Any trial that would be referred to as ‘‘phase 0’’ meets the definition of a phase 1 trial under FDA regulations (21 CFR312.21), and therefore, is generally, not required to be registered.
Some clinical trials are classified as phase 1/phase 2 trials (i.e., trials with characteristics of both phase 1and phase 2 studies). These clinical trials are not considered phase 1 studies and may be applicable drug clinical trials if they meet the other specified criteria, and therefore, may be required to be registered.
How do I determine if a device is FDA-regulated and requires registration on ClinicalTrials.gov?
There are several FDA and other websites that can assist you with determining if your device is FDA regulated. Please see below:
- FDA Device Classification Panels: https://www.fda.gov/medical-devices/classify-your-medical-device/device-classification-panels.
- Registration of a medical device: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm. This will let you know if it's a registered device or not (i.e. already on the market and what is the intent and indication for use).
- Workflow to make determination: http://regardd.org/devices/is-my-study-exempt.
- Product classification: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm. This will let you know if the level of control necessary to assure the safety and effectiveness of the device as well as other regulatory information.
- Lastly, FDA's Device Advice provides comprehensive regulatory assistance if needed in determinations. They are available by email at DICE@fda.hhs.gov and by phone at (800) 638-2041 and (301) 796-7100.
What happens to a ClinicalTrials.gov study record when the PI leaves UC San Diego?
When a Principal Investigator (PI) leaves UC San Diego, one of the things that is often overlooked is the ClinicalTrials.gov record. For studies where the PI is listed as the Responsible Party and is leaving UC San Diego, there are two options depending on whether the study will remain at UCSD or transfer with the PI:
- If the research will stay at UC San Diego and continue under a new PI, the new PI must be assigned to the ClinicalTrials.gov study record and the study status must be updated within 30 days after the change has been made. To update the record, the PI will need to send an email with the following information to the Research Compliance and Integrity Office (RCI) at ctgov@ucsd.edu who will update the record(s) on ClinicalTrials.gov:
- The new PI name
- Study status (recruiting, active but not recruiting, completed, etc.)
- The new IRB number (if a new IRB number has been assigned)
- If the research will continue at a new institution with the same PI, the ClinicalTrials.gov record can be transferred to the new institution. The PI must email RCI at ctgov@ucsd.edu and request the record transfer at least 30 days prior to transferring the study.
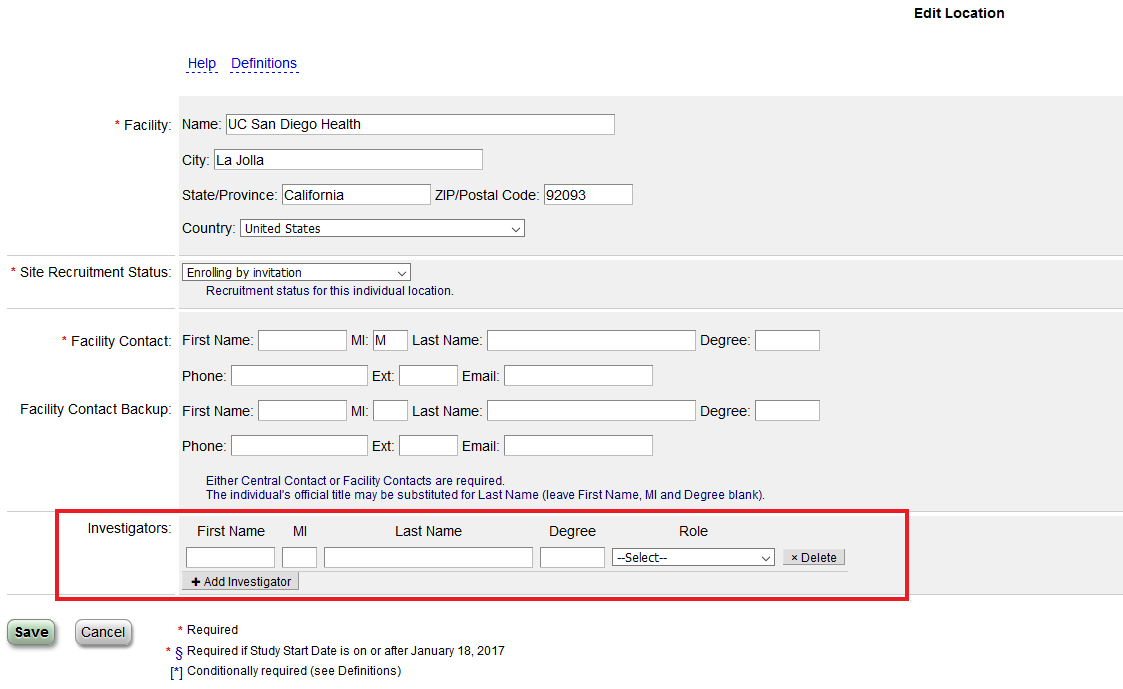
Can a Co-Investigator be added to the study record?
Yes, under the protocol section in the “Locations/Contacts” section you can add a site Principal Investigator or Sub-Investigator.
Expand section PROTOCOL REGISTRATION AND RESULTS SYSTEM (PRS)
What is the PRS?
The PRS, Protocol Registration and Results System, is the data entry and review system used to submit study data to the team that manages ClinicalTrials.gov. PRS is where Responsible Parties (RPs) register their studies, submit summary results information, and communicate with the ClinicalTrials.gov team. All information posted on the PRS will be publically published on the ClinicalTrials.gov webpage.
Is there a place I can test out entering data into the Protocol Registration and Results system (PRS)?
If you would like to explore the PRS and how to operate the system, you can use the “PRS Test System.” https://prstest.clinicaltrials.gov/. Please note you cannot transfer anything out of the test PRS system and import it into the regular PRS system. You will need a user account, to obtain a new user account, email ctgov@ucsd.edu.
What are the steps of using the Protocol Registration and Results System (PRS) system?
- Log in to PRS
You will need to log in UCSD’s Protocol Registration and Results System (PRS). For a new user account email ctgov@ucsd.edu.
- Create a new record
The person who creates the record becomes the Record Owner. All email messages about the record will be sent to this person. This person can be different from the Responsible Party.
- Enter (or edit) study information
Provide information summarizing the study protocol, including the Brief Title, Study Type, Outcome Measures, Arms and Interventions, Eligibility Criteria, Contacts, and Study Site Locations. See the Protocol Registration Data Element Definitions and the Expanded Access Data Element Definitions for a complete list of required and optional data elements.
- Submit the study record for PRS Review
After all study information is entered, the person entering the information (Record Owner) clicks on “Entry Complete.” The Responsible Party for the study clicks on “Approve” to accept the content and then the Responsible Party clicks on “Release” to submit the record for review by Protocol Registration and Results System (PRS) Staff.
- PRS Staff review the record
After the Responsible Party releases the record, PRS Staff review it for apparent errors, deficiencies, and/or inconsistencies. If PRS Staff find any potential issues with the record, they will add comments to the record and send an email notification to the Record Owner and Responsible Party. The user must log in to PRS to view the comments, and then make edits to the study record to address the comments and resubmits the record for PRS Review, using the same procedures described in steps three and four.
- Record is registered and posted
Once the study record passes PRS Review, an email notification will be sent with the ClinicalTrials.gov Identifier (NCT number), indicating that the study is registered. Generally, within two business days of registration, the system will post the record on the ClinicalTrials.gov website. Once registered, a study record becomes a permanent part of ClinicalTrials.gov and cannot be deleted.
- Keep record up to date
The study record needs to be verified and/or updated at least once a year (as described in steps three and four), with some data elements requiring more rapid updates, until the study is completed and/or the PRS Review process has concluded for submitted results information.
- Add results
U.S. regulations requires some studies to submit results to ClinicalTrials.gov. Generally, results must be submitted within one year of the Primary Completion Date.
Expand section PROBLEM RECORDS
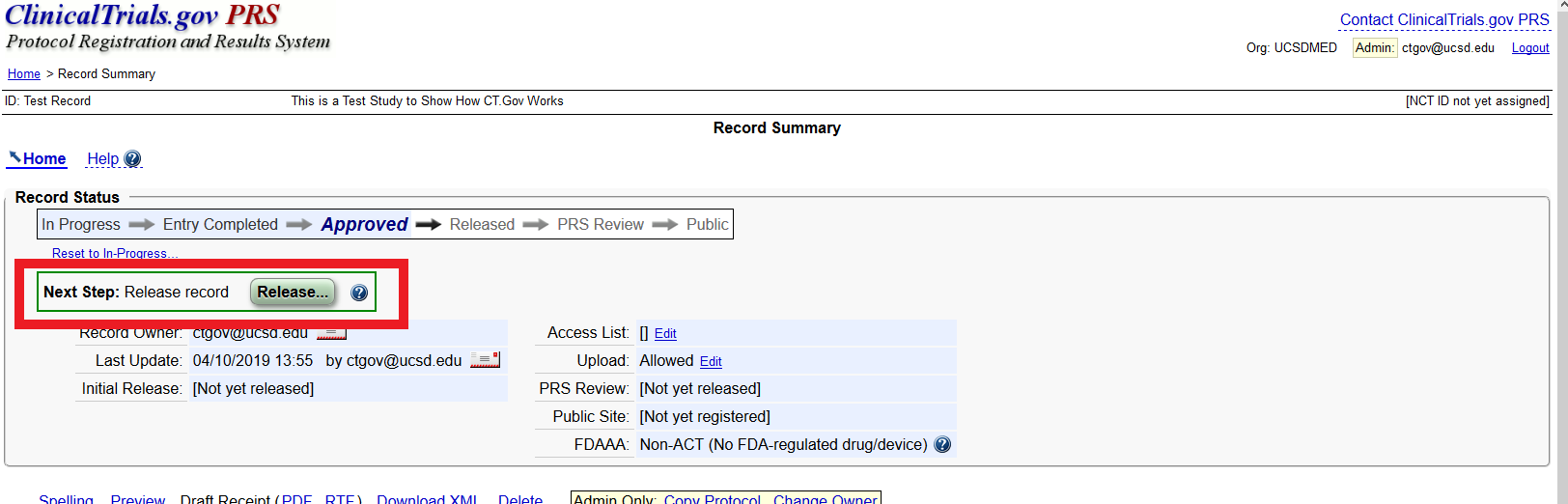
I approved my study record but the Protocol Registration and Results System (PRS) still says there is a problem with my record, what do I do?
A record must be marked as completed, approved, and released before it can be reviewed by PRS staff and made public (or updated) on the ClinicalTrials.gov website. If you are the Record Owner, you must click “Entry Complete” and then the record will be sent to the Responsible Party for next steps to “Approve” and “Release” the record. If you are the Responsible Party, you must both “Approve” and “Release” the record.
To mark the record “Entry Complete,” or to “Approve” or “Release” the record click the green box labeled “Approve” or “Release” in the “Next Step” Section of ClinicalTrials.gov.
ClinicalTrials.gov says my record has a problem, what does this mean?
- Record Has Errors: The study record has one or more error messages. The record must be modified to address the errors, and then the record must be “Marked as Completed,” “Approved,” and “Released.”
- Entry Not Complete: The record has been created or modified, but the Record Owner never marked the “Entry Complete” button. The record must be “Marked as Completed,” “Approved,” and “Released.”
- Not Recently Updated: The record has not been recently updated, a record must be updated at least one every 12 months. The record must be reviewed and updated as necessary then the record must be “Marked as Completed,” “Approved,” and “Released.”
- Pending Protocol Registration and Results System (PRS) Review: Potential issues were identified during the PRS review and the record must be modified to address the comments, and then the record must be “Marked as Completed,” “Approved,” and “Released.”
- Never Released: A record has been created, but has not been released for Protocol Registration and Results System (PRS) review. The record should be reviewed to determine if it should be posted on ClinicalTrials.gov. If yes, then the record must be “Marked as Completed,” “Approved,” and “Released.”
- Update Not Released: A record that has been registered and posted on ClinicalTrials.gov has been updated, but the update has not been released. The record must be reviewed and then the record must be “Marked as Completed,” “Approved,” and “Released.”
- Ready for Review and Approval: A user has marked a record as “Entry Complete” following the intitial data entry or modification of the record. The record is ready to be approved and and released for Protocol Registration and Results System (PRS) staff review. The record must be reviewed, and edited as needed, and then the record must be “Marked as Completed,” “Approved,” and “Released.”
My study record has a “Late Results—per FDAAA” problem, what does this mean?
This problem means this record has mandatory results reporting one year after the Primary Completion Date, and these results were never posted to ClinicalTrials.gov. First, determine if (1) results are required to be submitted or (2) if you can submit a certification or extension request to delay the results submission.
Often times, other study team members or organizational members may be more appropriate to enter the results data. Make sure they have user access and give them notice of the past due problem on the study record. Once complete have that person mark “Entry Complete.” The Responsible Party will then review and edit the record as needed, and select “approve” and “release” to submit the record to ClinicalTrials.gov for Protocol Registration and Results System (PRS) Review.
My study record has an “Incomplete Results—per FDAAA” problem, what does this mean?
This problem means this record appears to be missing secondary outcome measure results information.
Often times, other study team members or organizational members may be more appropriate to enter the results data. Make sure they have user access and give them notice of the past due problem on the study record. Once complete have that person mark “Entry Complete.” The Responsible Party will then review and edit the record as needed, and select “approve” and “release” to submit the record to ClinicalTrials.gov for Protocol Registration and Results System (PRS) Review.
Expand section PROTOCOL REGISTATION AND RESULTS SYSTEM (PRS) REVIEW AND REVIEWER COMMENTS
How long will the Protocol Registration and Results System (PRS) review take?
After you have registered your study, the PRS review may take approximately 2 to 5 business days. The PRS review of records with results information may take up to 30 days.
How do I view the Protocol Registration and Results System (PRS) comments?
A flag will appear next to each section and module of the study record that has at least one PRS review comment to be addressed. To read the comments, select the “Review Comments" box in the section.
To view all PRS Review Comments ever received on a record, select the “Review History” link on the “Record Summary” page. Then, select “Review Comments” for the version of the record you would like to see.
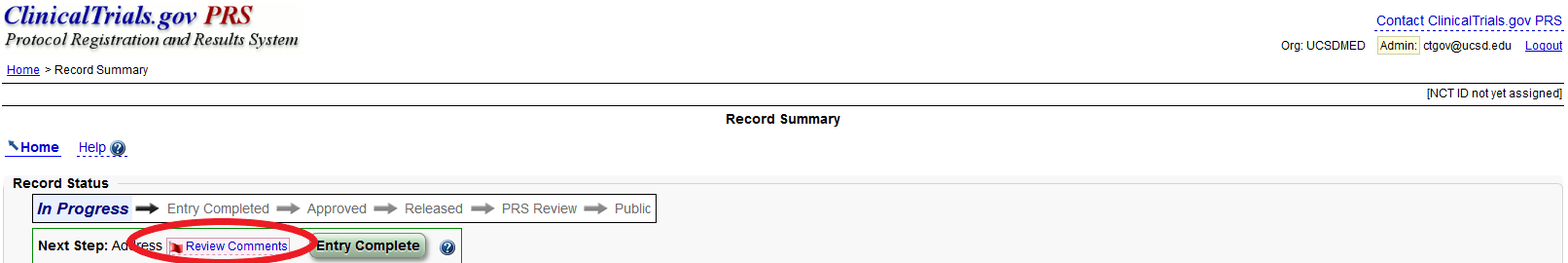
NOTE: When you click review comments, a new webpage will populate. You cannot edit the new webpage, you can only edit in the original screen.
How long do I have to respond to the Reviewer’s comments when they complete their Protocol Registration and Results System (PRS) review?
Registration: The Responsible Party must ensure “Major Comments” are addressed within 15 calendar days of the date on which PRS Staff sent the notification.
Results: The Responsible Party must ensure “Major Comments” are addressed within 25 calendar days of the date on which PRS Staff sent the notification.
The “Corrections Expected Date” is indicated in the Record Status box on the Record Summary page.
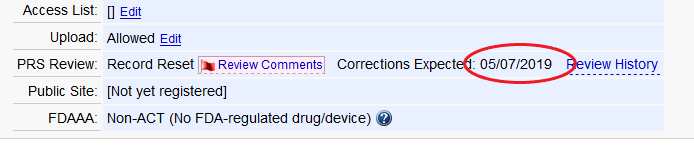
A Protocol Registration and Results System (PRS) reviewer just completed reviewing my record and there are comments. What do these mean and what do I do to fix them?
If PRS Staff identify potential issues with the study record, they will add reviewer comments to your record. The comments must be reviewed and/or addressed as necessary. Comments are identified as:
Major Comments: These must be corrected or addressed. A complete list of major comments that may be provided is available on ClinicalTrials.gov Major Comments page.
Advisory Comments: These should be addressed to improve the clarity of the record.
When the PRS staff identify potential issues during the review, an email notification is sent to the Record Owner with instructions for viewing the PRS Review Comments. Others receiving the email notification include the last user to update the record and the Responsible Party.
What is the difference between an Error, Warning and Note on the Protocol Registration and Results System (PRS) system?
The PRS will automatically detect some data entry issues when you save each module, and alert you when there are possible problems. Errors or incomplete information in one module may create data validation issues in another module. You may need to correct or add information in different modules to address all validation messages on your record.
ERROR messages indicate serious issues that must be addressed, and prevent the record from being released for PRS review. e.g., “Primary Completion Date should not be before the study start date.”
WARNING messages indicate potentially serious issues that should be reviewed and addressed as needed, usually indicates data has been omitted that is required by the regulations, and prevent the record from being released. e.g., “The IND serial number has not been entered.”
NOTE messages indicate potential issues that should be reviewed and addressed as needed. It does not prevent the record from being released, however a PRS reviewer will review the note. e.g., “A title this short may not be sufficiently descriptive.”
How do I know what the Protocol Registration and Results System (PRS) Reviewers are looking at?
PRS Staff rely on two sets of criteria when reviewing summary protocol and results information in study records:
- ClinicalTrials.gov Protocol Review Criteria
- ClinicalTrials.gov Results Review Criteria
It is strongly recommended that you check these criteria against your study record.
Expand section POSTING RESULTS
How long will it take me and my study team to upload the results data?
All studies have various amounts of results data that must be analyzed and published, but the initial entry of summary results information into Protocol Registration and Results System (PRS) can take as long as 40 hours. Please plan accordingly.
When does the obligation to maintain the study record end?
The study record’s “Protocol Section” requires regular updates as the study progresses. As the summary results information is reported after the end of the study, in most cases, there is no requirement to update the record once all comments raised during the Protocol Registration and Results System (PRS) review process are satisfactorily addressed.
All researchers are required to submit summary results information no later than 12 months after the primary completion date, even if their study remains ongoing – for example, data collection is ongoing for secondary outcome measures or adverse event information. However, there are exceptions to this requirement:
If a researcher discovers an error in the record, the researcher must correct it in PRS within 15 days for protocol registration and 25 days for summary results information.
If a researcher submitted a partial result, the researcher may be required to submit additional partial results and final summary results within 12 months of the actual study completion date.
Once results are published, a researcher may wish to edit the record to link to the publication. The same holds for conference presentations, which may be appropriately referenced in the record.
What happens if I did not collect any data regarding one of the primary or secondary outcome measures I entered during registration?
Often times studies change course, and the outcome measures that were first chosen at the beginning of the study are no longer the same outcome measures as the end of the study. When entering results, the information entered in during the registration process in the “Protocol Section” will be automatically carried over to the “Results Section.” Therefore, it is critical to ensure that the study record’s “Protocol Section” is accurate and up-to-date. Before adding or removing an outcome measures in “Results Section,” make sure you update the “Protocol Section.” The Protocol Registration and Review System (PRS) reviewer will be comparing the Primary and Secondary outcome measures in the Results Section, with those listed in the Protocol Section, and they will compare the Outcome Measures with those listed in the protocol uploaded under the Documents Section.
Can I ever delay posting my results?
A certification for the delayed submission of results information or request for an extension for good cause can be submitted via the Protocol Registration and Results System (PRS). A responsible party may delay the submission of results information by submitting a certification that one of the two following conditions has been met:
- Certify Initial Approval: The ACT reached its primary completion date on or after January 18, 2017, and before the drug, biological, or device product is initially approved, licensed, or cleared by FDA for any use (referred to on ClinicalTrials.gov as "certify initial approval").
- The earlier of the date that is 30 days after the date that:
- The drug, biological, or device product is approved, licensed, or cleared by FDA for any use that was studied in the ACT
- The application or premarket notification is withdrawn without resubmission for not less than 210 days
- Alternatively; 2 years after the date that a certification is submitted, if neither of the events listed above has occurred by that time.
2. Certify New Use: The ACT studies a new use of an FDA-approved drug, biological, or device product (that is, a use not included in the labeling), and the manufacturer of the drug, biological, or device product is the sponsor of the trial and has filed or will file within 1 year an application to FDA for approval or clearance of that use (referred to on ClinicalTrials.gov as "certify new use").
Results deadline:
- The earlier of the date that is 30 days after the date that:
- The new use of the drug, biological or device product is approved, licensed, or cleared by FDA;
- FDA issues a letter ending the regulatory review cycle for the application or submission for the new use of the drug, biological, or device product, such as a complete response letter, or
- The application or premarket notification for the new use is withdrawn without resubmission for no less than 210 days
- Alternatively; 2 years after the date that a certification is submitted, if none of the events listed above has occurred by that time.
What study documents are required to be uploaded to ClinicalTrials.gov and when should I upload them?
Study Protocol and Statistical Analysis Plan: The study protocol and statistical analysis plan are required for studies if they have a Primary Completion Date on or after 18 January 2017. The protocol must include a list of all prior Institutional Review Board (IRB)-approved versions and/or amendments. The cover sheet must identify the study’s national clinical trial (NCT) number. The protocol and the statistical analysis plan can be in the same document and just uploaded once.
Informed Consent Forms (ICF): Posting Informed Consent Forms (ICF) is required by the Common Rule, and the Office for Human Research Protections (OHRP) has identified two publicly available federal websites that will satisfy the posting requirement: a docket folder on Regulations.gov (Docket ID: HHS-OPHS-2018-0021) and in the study record on ClinicalTrials.gov. When using ClinicalTrials.gov to post the ICF, it must be the final version of the legal document that has been approved by the IRB, and contain a cover page that includes: (1) Official Study Title, (2) National Clinical Trial (NCT) identifier number (if available), and (3) Document Date.
Suggested best practice: load all documents within 30 days after the last day of data collection.
For more information, review our Factsheet: ClinicalTrials.gov Instructions for Uploading Study Documents and Redaction Guide.
Do I need to post the informed consent form?
The revised Common Rule requires that an IRB-approved version of an informed consent form be posted on a public federal website for all clinical trial conducted or supported by a Common Rule department or agency. This must be done after enrollment ends and within 60 days of the last study visit by any subject.
- You can upload an IRB-approved version of the form to the ClinicalTrials.gov study record. Note that ClinicalTrials.gov does not accept non-English documents.
- Be sure to follow the Protocol Registration and Results System (PRS) instructions for document uploads at https://prsinfo.clinicaltrials.gov/results_definitions.html#DocumentUpload
The Common Rule (45 CFR §46.116(h)) and NIH Notice (NOT-OD-19-050) differ in the date the Informed Consent Form should be posted, which should I follow?
The Common Rule (45 CFR §46.116(h)) reads, “(3) The informed consent form must be posted on the Federal website after the clinical trial is closed to recruitment and no later than 60 days after the last study visit by any subject, as required by the protocol”, however the NIH Notice (NOT-OD-19-050) reads “For NIH-funded or supported clinical trials, informed consent documents must be posted on a public federal website after recruitment closes and no later than 60 days after the last study visit.” Although the text in the NIH Notice excludes the phrase, “by any subject”, the requirement is the same between the Common Rule and NIH, i.e. post the consent form after recruitment closes for your study and no later than 60 days after the last study visit.
What if the study is old and I don't have the data anymore?
If you receive an email from RCI that alerts you that an older study must have the results posted and you are unsure of where the study data is and don't know how to enter results with the little information you may have, enter whatever data can be found. For instance, the number of participants, age and sex may be known, or perhaps some outcome measure or adverse event data were recorded. In other places where the data cannot be located, it may be possible to enter zero analyzed, with an explanation that the data was not accessible and all efforts have been made to locate the data.
RCI can also provide assistance. Contact ctgov@ucsd.edu and RCI will work with you and find the best way to enter the data you have located.
What if there is Personal Identifiable Information, trade secrets, or confidential commercial information on the study documents. Do I still have to upload the documents?
Yes, some Protocols may include information that is necessary to redact before making public. Such information may include personal identifiable information, trade secrets, and confidential information (e.g., exploratory endpoints).
For more information, review our Factsheet: ClinicalTrials.gov Instructions for Uploading Study Documents and Redaction Guide.
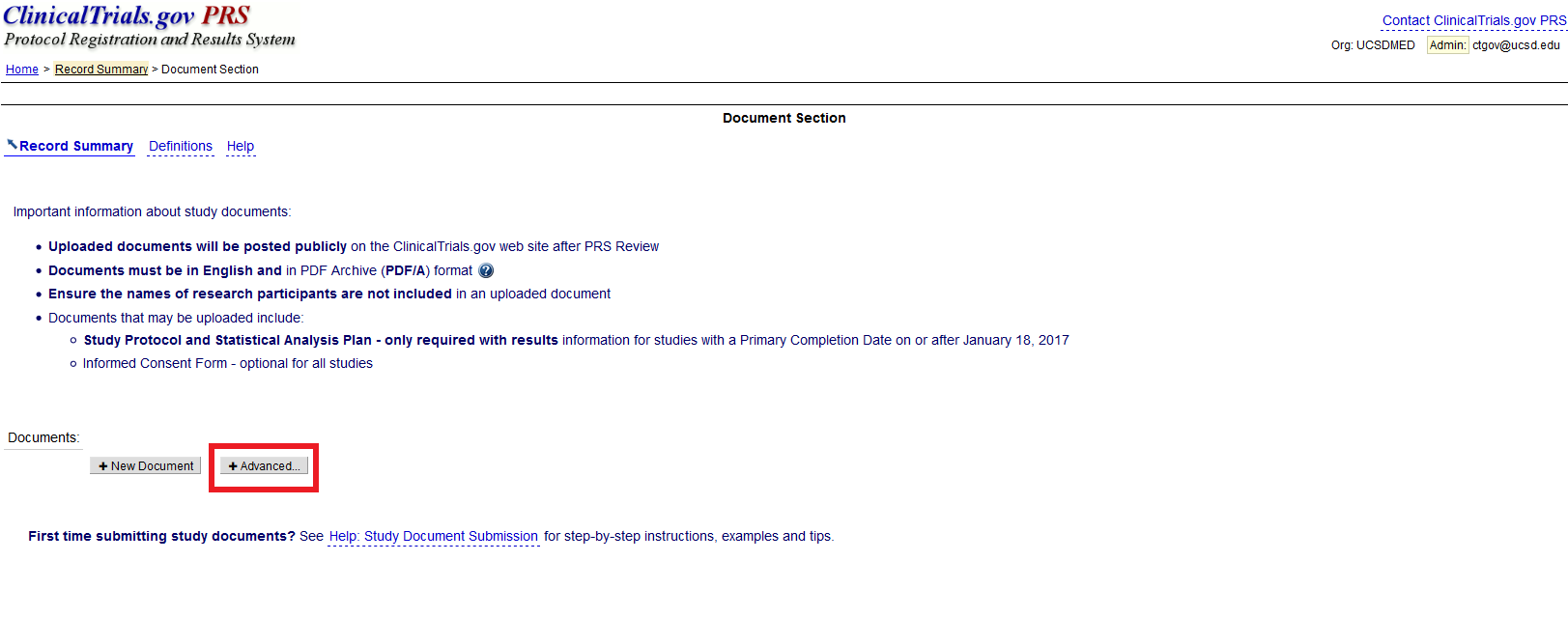
Can I upload more than one document at a time on ClinicalTrials.gov?
There is also a “+ Advanced” button. This button permits users to upload more than one document at a time – for example, the study protocol, statistical analysis plan, and ICF.
What is “Voluntary Reporting?”
Those who are not required to register their studies or publish their results on ClinicalTrials.gov may do so, even if not required, it is called “voluntary reporting.” However, if ones choses to register and report their results, they must go through the same process as those required to post. The Protocol
Registration and Review System (PRS) review will still occur, and the record will still have to be updated and monitored.
What is an “Extension for Good Cause?” and when is the deadline to submit one?
The Director of the National Institute of Health (NIH) may extend the deadline for submission of results information for an Applicable Clinical Trial (ACT) if the responsible party submits a written request that demonstrates good cause for the extension and provides an estimated date on which the results information will be submitted. The Director will review and notify the responsible party as to whether the requested extension of the deadline for submitting results information demonstrates good cause and has been granted. Before this notification is issued, the processing of such requests by NIH does not mean that the NIH Director has determined the request demonstrates good cause. In general, pending publication would not be considered good cause for an extension.
As of January 25, 2022, the PRS no longer permits good cause extension requests submitted late. A late submission is one that is requested on or after the deadline of results posting for Applicable Clinical Trials. An extension for good cause may still be requested prior to the date that results would be due.
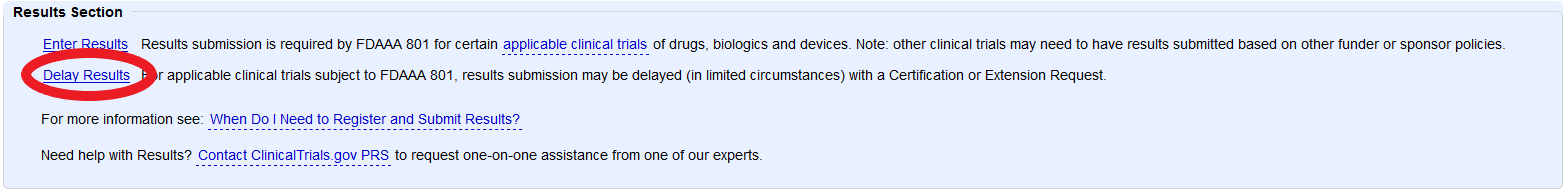
How do I request certification of delay or a good faith extension?
Under the “Results Section” click Delay Results and enter certification or extension information. When done, mark “Entry Complete.” The Responsible Party will have to review and edit the record as needed, and mark “approve” and then “release” to submit the record to ClinicalTrials.gov for The Protocol Registration and Review System (PRS) Review.
What is the deadline for submitting a certification for delayed submission of results information?
The responsible party must submit the extension request via the ClinicalTrials.gov Protocol Registration and Results System (PRS) prior to the date (i.e., the day before) that results information would otherwise be due according to 42 CFR 11.44(a) through (f). The extension request must include a description of the reasons that the responsible party believes constitute good cause to justify an extension and an estimated date on which the results information will be submitted, with sufficient detail to allow for evaluation of the request.
As of January 25, 2022, the PRS no longer permits responsible parties to submit good cause extension requests late (i.e., on or after the results information submission deadline) for ACTs with a primary completion date on or after January 18, 2017. This change aligns the PRS's automated validation rules with the regulatory requirements in 42 CFR 11.44(e). Prior to January 25, 2022, a responsible party could have submitted a late good cause extension request.
Failure to submit required results information is a prohibited act under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. 331(jj)(2), for which FDA could pursue civil monetary penalties under 21 U.S.C. 333(f)(3) against the ACT's responsible party. For an ACT for which an NIH or FDA grant recipient is the responsible party, failure to submit required results information could result in NIH or FDA, as applicable, not releasing remaining funding for a grant or funding for a future grant, pursuant to section 402(j)(5)(A) of the Public Health Service Act.
As of January 25, 2022, the PRS no longer permits responsible parties to submit good cause extension requests late (i.e., on or after the results information submission deadline) for ACTs with a primary completion date on or after January 18, 2017. This change aligns the PRS's automated validation rules with the regulatory requirements in 42 CFR 11.44(e). Prior to January 25, 2022, a responsible party could have submitted a late good cause extension request.
Failure to submit required results information is a prohibited act under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. 331(jj)(2), for which FDA could pursue civil monetary penalties under 21 U.S.C. 333(f)(3) against the ACT's responsible party. For an ACT for which an NIH or FDA grant recipient is the responsible party, failure to submit required results information could result in NIH or FDA, as applicable, not releasing remaining funding for a grant or funding for a future grant, pursuant to section 402(j)(5)(A) of the Public Health Service Act.
Can I include the study data analysis in the study timeline?
Data analysis is not to be included in the study timeline. The study is completed when the team is finished collecting data from the study subjects (i.e., all the subject visits are done). The primary completion date is the last date data was collected from the subject for the primary outcome measures. The study completion date is the last date data was collected for all the other pre-specified outcome measures. At the point of study completion, results must be posted to CinicalTrials.gov within 12 months.
I have received an email from CT.gov that says my results are now required to be posted, but previously they were not required. What does this mean?
Federal Court Decision decision, Seife et al. v. HHS et al., 18-cv-11462 (NRB) (S.D.N.Y. Feb. 24, 2020), requires submission of results information for an "applicable clinical trial" (ACT) that was initiated after September 27, 2007, or that was ongoing as of December 26, 2007, if the ACT studies a product that is approved, licensed, or cleared by the FDA at any time, including after the ACT's primary completion date.
If the results submission deadline has already passed, submit the results information as soon as possible.
If the results submission deadline has already passed for an ACT affected by the Federal Court decision in Seife et al. v. HHS et al., 18-cv-11462 (NRB) (S.D.N.Y. Feb. 24, 2020), what is the deadline for submitting results information?
Responsible parties for such ACTs should submit the results information specified in section 402(j)(3)(C) and section 402(j)(3)(I) of the PHS Act as soon as possible.
I have submitted a publication to a journal and the journal has placed an embargo on the publication. Can I wait until after the embargo is lifted to post the study results (that are included in the publication) to ClinicalTrials.gov?
No, ClinicalTrials.gov does not provide extensions or waive “late results” findings for a publication that has been submitted to a journal and has been embargoed. The requirements for submission to ClinicalTrials.gov are separate from those related to publishing. ClinicalTrials.gov requires results to be posted to ClinicalTrials.gov no later than one year after the study's Primary Completion Date, as described in 42 CFR 11.44(a). Fines and penalties for late results may include civil monetary penalties of $13,237 per a day the study is in noncompliance, loss of Health and Human Service (HHS) funding to the study and/or UC San Diego and/or public notices of non-compliance.
It is the responsibility of each Responsible Party to check with the journal to determine the journal’s individual policy. Please note that the ICMJE, as well as several separate Journal editorials, have made clear that submission of results to ClinicalTrials.gov will not constitute "prior publication" for the purposes of manuscript consideration.
How do I submit results for a trial that was terminated and had no participants enrolled? Alternatively, how do I submit results for a trial that was terminated and participants were enrolled, but no data was collected for one or more Outcome Measures?
If no participants were enrolled in the trial, set the Overall Recruitment Status to Withdrawn, and no results information will need to be submitted.
If participants were enrolled before trial termination but no data were collected for one or more Outcome Measures, specify zero ("0") in the relevant Number of Participants Analyzed data element and leave the data fields blank for each Arm/Group. In this case, use the Analysis Population Description data element to explain that the trial was terminated before the outcome measure data were collected and, if appropriate, provide additional details in the Limitations and Caveats module. All other required results information collected for the enrolled participants will need to be submitted in each results module even though no data were collected for one or more Outcome Measures.Expand section POSTING INFORMED CONSENT DOCUMENTS
Which studies must post an Informed Consent document onto ClinicalTrials.gov?
Per 45 CFR 46.116(h), there are two categories of clinical trials that require ICFs to be posted:
- Category 1: Nonexempt clinical trials, that are conducted or supported by HHS, initially approved by an IRB on or after January 21, 2019.
- Category 2: Nonexempt clinical trials conducted or supported by HHS, initially approved by an IRB before January 21, 2019, that continue on or after January 21, 2019, and for which the following are true:
- An institution transitions a clinical trial to comply with the 2018 Requirements in compliance with the transition provision; and
- The transition determination was documented and dated by the IRB or institution before the clinical trial is closed to recruitment and 60 or fewer days before the last protocol-required study visit by any subject enrolled in the protocol
When must a consent form be posted for clinical trials initially approved on or after January 21, 2019?
A consent form must be posted to ClinicalTrials.gov after the clinical trial is closed to recruitment, and no later than 60 days after the last study visit by any subject, as required by the protocol. If a consent form is posted before a clinical trial closes recruitment, it would have to be re-posted after the clinical trial closes recruitment.
What is considered the last study visit by any subject, as required by the protocol?
The last study visit is the last visit that would not have happened as part of routine clinical care. This visit is any visit that a subject has with the research team including in-person visits, filling out a survey, or a final telephone conversation, that is required by the protocol.
Can information on the consent form be redacted?
Redacted forms may only be posted only if the federal department or agency conducting or supporting a trial has permitted or required redactions.
Will uploading a consent form to one ClinicalTrials.gov within the required time frame satisfy the posting requirements even though that uploaded form may not be visible on the website until a later date?
Yes. Uploading and submitting the ICF to ClinicalTrials.gov will satisfy the posting requirements.
If there is more than one IRB-approved version of a consent form, how many must be posted?
Only one IRB-approved consent form must be posted.
Should an unsigned consent form be posted?
Yes. No personally identifiable information or other patient information should be on the form.
If an HHS-supported cooperative research clinical trial involves multiple sites that close recruitment at different times, when is the earliest that a consent form can be posted in order to satisfy the posting requirements?
The earliest that a consent form may be posted is when all sites have closed to recruitment.
If a short form written consent procedure is used, must it still be posted to ClinicalTrials.gov?
Yes. One copy of the short form and the study summary that was presented orally to subjects during subject enrollment must be posted.
Expand section FDA PRE-NOTICE AND NOTICE OF NONCOMPLIANCE LETTERS
What is a Food and Drug Administration (FDA) Preliminary Notice of Noncompliance (Pre-Notice) letter?
A Preliminary Notice of Noncompliance (Pre-Notice) Letter describes the potential violation and requests that the responsible party take the necessary actions to address the potential violation within 30 calendar days after receiving the letter. After 30 calendar days from receipt of a Pre-Notice Letter, the Food and Drug Administration (FDA) will conduct a further review and assessment of the clinical trial information submitted to ClinicalTrials.gov (and other relevant information available to FDA) to determine if regulatory action is required, inclusive of the issuance of a Notice of Noncompliance, civil money penalties, an injunction, and/or criminal prosecution.
Have any Food and Drug Administration (FDA) Pre-Notice letters been issued?
Yes, the Food and Drug Administration (FDA) has issued pre-notice letters to Responsible Parties (RP).
What does a Food and Drug Administration (FDA) Pre-Notice Letter look like?
Click here to view an actual sample Food and Drug Administration (FDA) Pre-Notice Letter.
What is a Notice of Noncompliance?
The FDA intends to post all Notices of Noncompliance on the agency’s website and to transmit the Notices of Noncompliance to the National Institutes of Health (NIH), so that NIH can include the statutorily-required notice regarding noncompliance in the ClinicalTrials.gov data bank. The Public Health Service (PHS) Act requires NIH to include in the data bank a notice that the responsible party is not in compliance with the law, the civil money penalties imposed for the violation, if any, and whether the violation has been corrected.
Will the letters go to the Principal Investigator (PI) or to the ClinicalTrials.gov Administrators?
Food and Drug Administration (FDA) obtains the Responsible Party’s (RP) contact information from the Protocol Registration and Results System (PRS). Note, for Principal Investigator (PI) initiated projects, UC San Diego requires that the PI be the RP. Therefore, it is important that the RP’s information in the PRS is accurate and current.
Is the Notice of Non-Compliance letter intended for Applicable Clinical Trials (ACTs) only or both ACTs and National Institutes of Health (NIH)-funded trials?
Food and Drug Administration’s (FDA) authority to issue a Notice of Noncompliance (under section 402(j)(5)(C)(ii)) applies to all applicable clinical trials regardless of how those trials are funded.
In addition, the FDA may issue a Notice of Noncompliance if it determines that a submitter failed to submit to the FDA the certification required by 402(j)(5)(B) of the Public Health Services (PHS) Act, or knowingly submitted a false certification. See the final guidance “Form FDA 3674-Certifications to Accompany Drug, Biological Product, and Device Applications/Submission.”
FDA also notes that for an applicable clinical trial for which a National Institutes of Health (NIH) or FDA grantee is the responsible party, failure to submit the required results information could result in the NIH or FDA (as applicable) not releasing the remaining funding for a grant or funding for a future grant, pursuant to section 402(j)(5)(A) of the PHS Act.
Expand section ADVERSE EVENTS
When do Adverse Events have to be reported?
Adverse events are not reported until the completion of the study when entering the results into ClinicalTrials.gov.
What Adverse Events have to be reported?
The adverse events module consists of three adverse event groups:
-
All-Cause Mortality: All deaths, due to any cause, that occurred during the study (required if Primary Completion Date is on or after January 18, 2017).
-
Serious Adverse Events: All serious adverse events that were collected during the study, whether or not they were anticipated or considered to be attributed or associated with the intervention.
-
Other (Not Including Serious) Adverse Events: Non-serious adverse events that were collected during the study, whether or not they were anticipated, and that occurred above a specified frequency threshold anywhere from 0% to 5% within any arm of the clinical study, grouped by organ system, with the number and frequency of such events by arm or comparison group of the clinical study.
For “Other Adverse Events”, what is the Frequency Threshold?
When entering the “other adverse events” the responsible party must identify the frequency threshold they are reporting. The frequency threshold can be anywhere from 0% to 5%. If the frequency threshold is specified as 5%, then any non-serious adverse event that occurred in more than 5% of participants in any of the arms must be reported. If the frequency threshold is specified as 0%, then any non-serious adverse event that occurred to any participants in any of the arms must be reported.
How do I determine the total number of “participants at risk” when reporting Adverse Events?
The total number of participants at risk will be the total number of participants that were enrolled in the study. The number of total number of participants at risk should match the enrollment number, if it does not match, explain the difference in the “adverse event reporting description.”
What is a "Serious Adverse Event?"
Serious adverse events include any adverse events that result in any of the following outcomes: death, a life-threatening adverse event, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant incapacity or substantial disruption of the ability to conduct normal functions, or a congenital anomaly/birth defect. Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious when, based upon appropriate medical judgment, they may jeopardize the participant and may require medical or surgical intervention to prevent one of the outcomes listed in this definition.
What does it mean that the Adverse Events have to be reported by organ system?
ClinicalTrials.gov requires you to assign each adverse event to a body or organ system. The choices are:
- Blood and Lymphatic System Disorders
- Cardiac Disorders
- Congenital, Familial and Genetic Disorders
- Ear and Labyrinth Disorders
- Endocrine Disorders
- Eye Disorders
- Gastrointestinal Disorders
- General Disorders
- Hepatobiliary Disorders
- Immune System Disorders
- Infections and Infestations
- Injury, Poisoning and Procedural Complications
- Investigations
- Metabolism and Nutrition Disorders
- Musculoskeletal and Connective Tissue Disorders
- Neoplasms Benign, Malignant and Unspecified (Including Cysts and Polyps)
- Nervous System Disorders
- Pregnancy, Puerperium and Perinatal Conditions
- Product Issues
- Psychiatric Disorders
- Renal and Urinary Disorders
- Reproductive System and Breast Disorders
- Respiratory, Thoracic and Mediastinal Disorders
- Skin and Subcutaneous Tissue Disorders
- Social Circumstances
- Surgical and Medical Procedures
- Vascular Disorders
Expand section COMPLIANCE
What are the consequences for noncompliance?
There are potential legal consequences for the Responsible Parties if they do not comply with the requirements to submit registration and results information on ClinicalTrials.gov. The consequences may include civil or criminal actions, civil monetary penalties, and grant funding implications. Below are some examples:
- Food and Drug Administration Amendments Act (FDAAA): Public notices of noncompliance and violations, withholding of federal funds, FDA sanctions, and civil monetary penalties of $13,237 per day.
- NIH: Suspension or termination of NIH grant or contract funding and consideration in future funding decisions.
- ICMJE: Refusal of publication in member medical journals following ICMJE policy.
Refer to the ClinicalTrials.gov Requirements Factsheet (PDF) for more information on the summary of requirements for different agencies.
What can I include for the NIH required Dissemination Plan?
The NIH requires there be a dissemination plan as part of the grant process. The plan must include the following:
- That the applicant will ensure that clinical trials under the award are registered and results information is submitted to ClinicalTrials.gov as outlined in the policy and according to the specific timelines stated in the policy;
- The informed consent documents for the clinical trial(s) will include a specific statement relating to posting of clinical trial information at ClinicalTrials.gov; and
- That the recipient institution has an internal policy in place to ensure that clinical trials registration and results reporting occur in compliance with policy requirements.
The following Factsheet may be provided to the agency or sponsor to address the institutional plan for compliance with the policy requirements.
Factsheet: UC San Diego ClinicalTrials.gov Requirements (PDF)
Does NIH verify ClinicalTrials.gov registration and results reporting?
All National Institutes of Health (NIH) funded clinical trials are required to be registered and have the results submitted on ClinicalTrials.gov per the “NIH Policy on Dissemination of NIH-Funded Clinical Trial Information.” The system validation in the electronic Research Administration (eRA) Human Subjects System (HSS) will result in an error for grant recipients upon submission of a Research Performance Progress Report (RPPR) when the clinical trial registration and/or results reporting are overdue.
- Registration: Grant recipients will receive a warning if they are not in compliance at 21 days after the enrollment of the first study participant. An error is then generated if the clinical trial registration is more than 30 days past this date. The registration error for RPPR is resolved when the trial is registered on ClinicalTrials.gov and the ClinicalTrials.gov identifier (NCT#) or the ClinicalTrials.gov registration receipt is submitted on the Human Subjects Clinical Trial Information (HSCT) form.
- Results reporting: Grant recipients receive an error if the results are overdue by more than 12 months after the clinical trial’s actual primary completion date. The results reporting error is resolved when results are submitted on ClinicalTrials.gov or by providing a ClinicalTrials.gov receipt for either a “Good Cause Extension" request or a “Certification of Delayed Submission of Results Information” on the HSCT form.
Grant recipients will receive an error at the time of RPPR submission, and the award will be prevented if the requirements are not met at the time the award per NOD-OD-22-008.
Is there a requirement to refer to ClinicalTrials.gov on the informed consent form?
Informed consent documents for applicable clinical trials must include the following statement that refers to the trial’s description on ClinicalTrials.gov:
‘‘A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time.’’
One component of FDA inspections is the registration of studies on ClinicalTrials.gov. The FDA inspector will determine whether the required statement referencing ClinicalTrials.gov is included when examining informed consent document(s). Please see the FDA Bioresearch Monitoring (BIMO) program guidelines for inspections for more information.
What are the FDA penalties related to ClinicalTrials.gov?
FDA recently issued final guidance on issuing Civil Monetary Penalties relating to the clinicaltrials.gov Data Bank. This guidance addresses FDA’s imposition of civil monetary penalties for failure to submit required clinical trial registration and results information.
A “responsible party” must submit registration and results information to the clinicaltrials.gov data bank for certain “applicable clinical trials.” (A responsible party is the “sponsor” of the clinical trial as defined in 21 C.F.R. 50.3, or the principal investigator if designated by the sponsor.) Additionally, a “submitter” of applications or submissions to the FDA (i.e., NDA, 510(k), PMA, HDE) is also required to certify to FDA that all requirements to submit information to clinicaltrials.gov have been met. The FDAAA authorizes the FDA to assess civil monetary penalties for failure to submit the required information to clinicaltrials.gov or to the FDA in certifications, or submitting false information to clinicaltrials.gov or in certifications.
How does a FDA Center intend to identify whether responsible parties have failed to submit required clinical trial registration and/or results information to the ClinicalTrials.gov data bank or submitted false or misleading information to the data bank, or whether submitters have failed to submit to FDA the certification required by section 402(j)(5)(B) of the PHS Act or knowingly submitted a false certification to FDA?
They intend to identify violations through evidence collection during Bioresearch Monitoring Program (BIMO) inspections. They may also identify violations based on evaluation of complaints received by FDA.
Under what circumstances may a FDA Center decide to seek civil money penalties against a responsible party or submitter?
If FDA believes a responsible party or submitter committed a violation, it will send a Preliminary Notice of Noncompliance (Pre-Notice) Letter, describing the potential violation, and will request that the recipient make any necessary corrections within 30 days. After 30 days, FDA will conduct a further review, and will issue a Notice of Noncompliance if it determines that a violation still exists. The recipient will then have 30 days to remedy noncompliance before FDA will seek civil monetary penalties.
In determining when to issue a Pre-Notice Letter, FDA intends to focus its attention to:
- Responsible parties who have failed to submit required information to clinicaltrials.gov for higher risk applicable clinical trials or applicable clinical trials of public health importance;
- Responsible parties or submitters for which there is a pattern of previous noncompliance with requirements to submit clinical trial information or certifications; and
What procedures apply when a Center seeks civil money penalties?
Actions are initiated when the FDA Center with principal jurisdiction files a Complaint and serves it on the responsible party or submitter. The respondent may respond by paying the penalty or filing an Answer. FDA regulations governing civil monetary hearings are found at 21 CFR Part 17.
What Civil Monetary Penalty amounts may be assessed for (1) failing to submit required clinical trial registration and/or results information to the ClinicalTrials.gov data bank, (2) submitting false or misleading information to the data bank, (3) failing to submit the required certification to FDA, or (4) knowingly submitting a false certification to FDA?
The statutory maximum penalties are not more than $10,000* for all violations adjudicated in a single proceeding and, if a violation is not corrected within 30 days following notifications of such violation, an additional Civil Monetary Penalty (CMP) of not more than $10,000* for each day that the violation continues after such period until the violation is corrected. In determining the amount of CMP under the relevant statutory limits, the following factors are considered: the nature, circumstances, extent, and gravity of the violation(s) and, with respect to the violator, ability to pay, effect on ability to continue to do business, any history of prior such violations, the degree of culpability and such other matters as justices may require.
*Amount adjusted annually to reflect inflation
Expand section RESOURCES
If I have a question, is there someone at ClinicalTrials.gov that can help me?
Researchers can contact the ClinicalTrials.gov team for assistance. Their support can be particularly helpful when it comes to complex study designs or issues with submitting summary results information into The Protocol Registration and Review System (PRS). In most cases, the ClinicalTrials.gov team provides guidance by email, but in some cases, they may also offer to set up a teleconference at a mutually convenient time to discuss study specifics and provide tailored guidance as to how to enter results into PRS. Email ClinicalTrials.gov at register@clinicaltrials.gov.
Note: A “Contact ClinicalTrials.gov PRS” link is available in the PRS.

Are there any tools to help me with registering my clinical trial and/or posting my results?
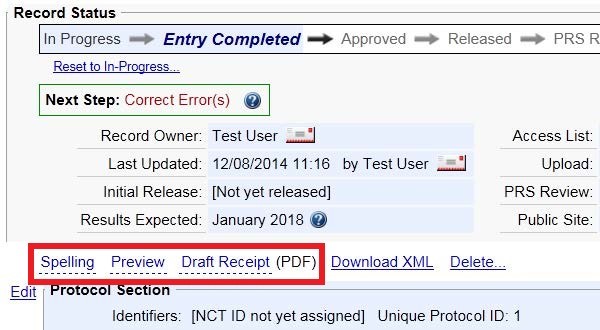
Spelling: The spelling option finds possible spelling errors and possible unexpanded acronyms. Only free-text data elements, such as Brief Title and Detailed Description, are checked. Some valid words, such as newer drug names, may not be recognized. Certain types of words, such as acronyms (all uppercase or mixed-case) and chemical names (combinations of letters and numbers), are not checked for spelling.
Preview: Gives you a rough approximation of how the Protocol Registration will appear on the ClinicalTrials.gov public web site.
Help: offers introductory information on the protocol data entry process.
Definitions: describes the definitions for protocol registration data elements
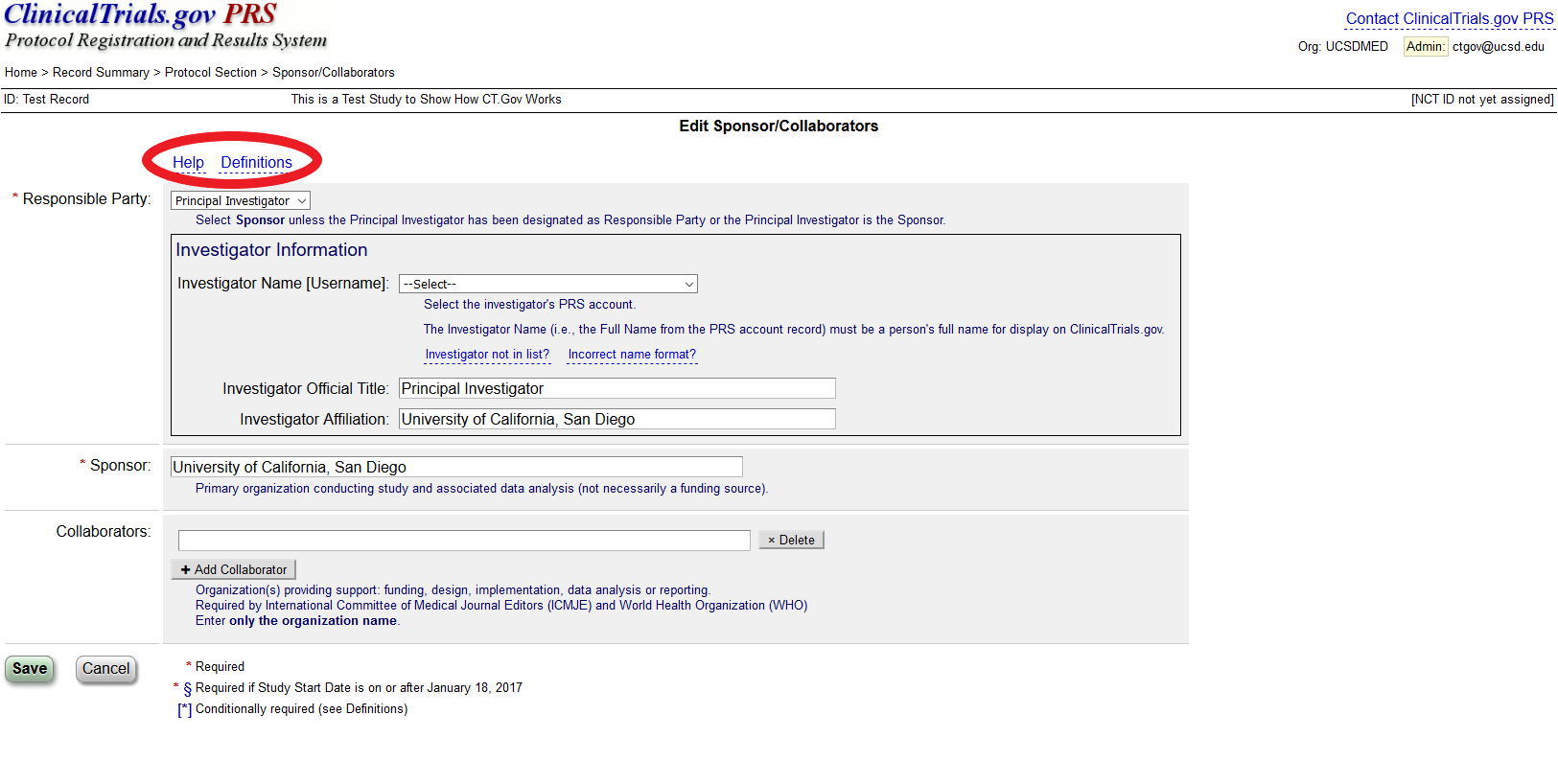
Are there scholarly publications about ClinicalTrials.gov?
Yes, please see the ClinicalTrials.gov Selected Publications page which contains links to scholarly publications regarding ClinicalTrials.gov.
Other tips for using ClinicalTrials.gov?
- Chrome and Firefox are more likely to let you “expand” text boxes to see more information.
- Use MS Word to create and edit these fields carefully, and then copy and paste into CT.gov fields.
- Do not use first or second person. Replace “I” and “we” with “the investigator”; replace “you” with “participants.”